

Болезнь накопления эфиров холестерина и болезнь Вольмана являются клиническими фенотипами единого патологического процесса — дефицита лизосомной кислой липазы (ДЛКЛ), являющейся наследственной болезнью накопления.
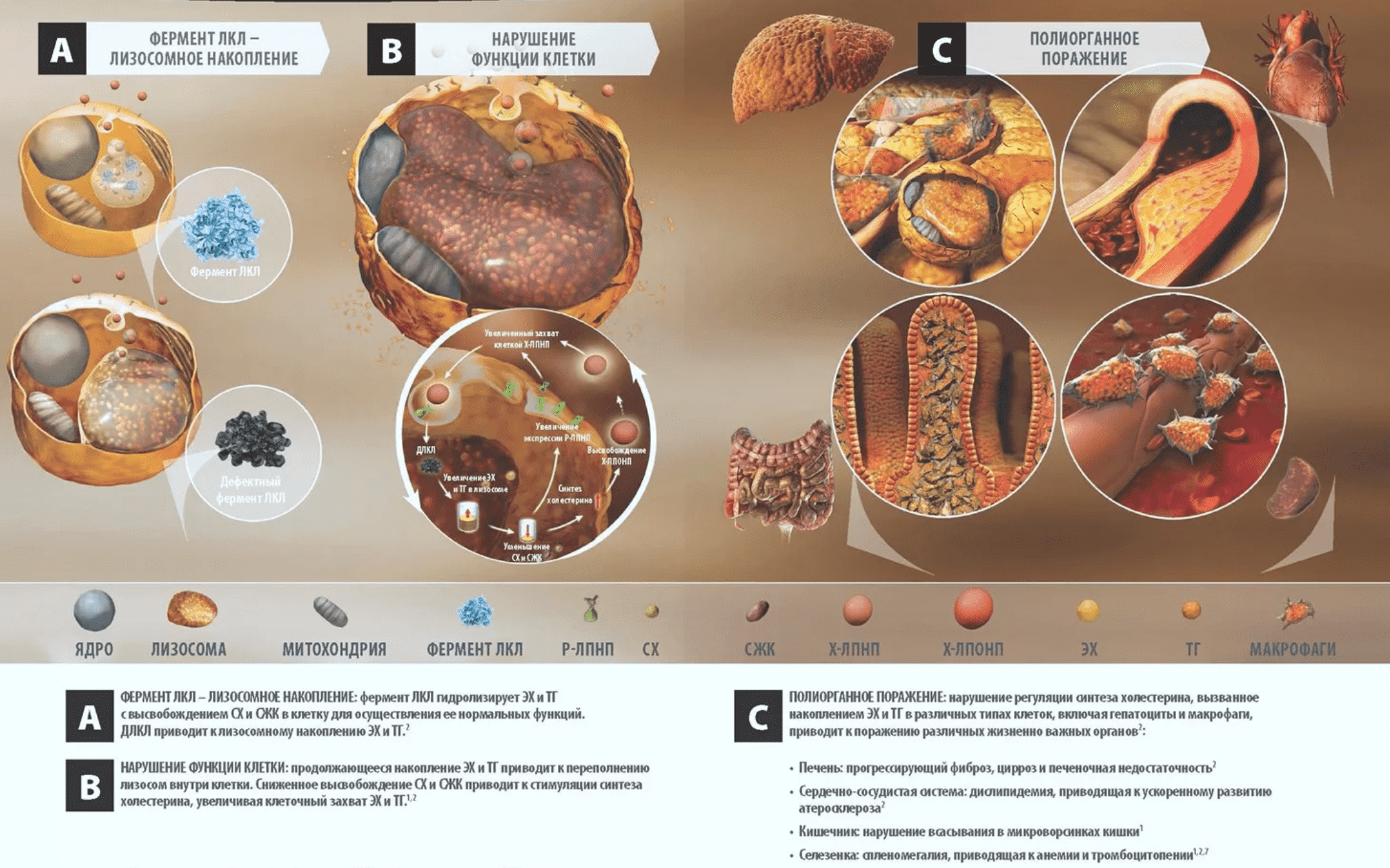
Вообще, лизосомная кислая липаза (ЛКЛ) представляет собой фермент клеточного метаболизма, обеспечивающий гидролиз эфиров холестерина и триглицеридов. Этот процесс является одним из самых важных в обеспечении клеток организма холестерином, принимающим участие в росте клетки и формировании клеточной мембраны. Врожденная мутация гена LIPA, ответственного за синтез ЛКЛ, приводит к значимому снижению активности данного фермента, нарушению метаболизма эфиров холестерина и триглицеридов и прогрессирующему накоплению их в клеточных лизосомах. Данный патологический процесс приводит к нарушению нормального функционирования клетки и повреждению различных органов и тканей. К основным органами-мишеням при ДЛКЛ относятся печень, селезенка, надпочечники, кишечник, стенки сосудов, однако и другие органы также могут вовлекаться в патологический процесс. Это происходит за счет поврежденных макрофагов, в метаболизме которых ЛКЛ играет ведущую роль.
Разнообразие клинических проявлений данной группы заболеваний обусловлено системным воздействием ДЛКЛ. Именно поэтому часто страдает своевременность точной диагностики.
Выделяют так называемую инфантильную форма ДЛКЛ, или болезнь Вольмана, характеризующуюся развитием в первые недели жизни ребенка тяжелой, быстропрогрессирующей полиорганной патологии, проявляющейся нарушением вскармливания, тошнотой, рвотой, симптомами мальабсорбции (нарушение всасывание нутриентов), задержкой роста и развития, а также исходно тяжелым повреждением печени. Эта форма наблюдается педиатрами, так как при ранних проявлениях ДЛКЛ прогноз крайне неблагоприятный, ибо медиана возраста смерти пациентов составляет 3,7 мес.
А вот такое состояние как болезнь накопления эфиров холестерина, наоборот, протекает зачастую скрыто (латентно), бессимптомно и является диагностической находкой при проведении рутинного осмотра и биохимического контроля крови, назначенных по иным причинам (диспансеризация, предоперационное обследование, например). При этой форме заболевания проявлениями болезни накопления эфиров холестерина являются симптомы повреждения печени (ее увеличение - гепатомегалия, повышение активности аланинаминотрансферазы (АЛТ) и аспартатаминотрансферазы (АСТ), ожирение печение - стеатоз, фиброз и даже цирротическая трансформация печени). Кроме того, могут наблюдаться увеличение селезенки (спленомегалия) и ее функции (симптомы гиперспленизма, например, анемия и тромбоцитопения), раннее развитие атеросклеротических поражений сосудов. Несмотря на «молчаливое», бессимптомное течение болезни, прогноз у пациентов с этой формой ДЛКЛ без лечения чаще всего неблагоприятный. Так, у 78% пациентов с ДЛКЛ выявляется фиброз и/или цирроз печени (по данным биопсии) с бессимптомным прогрессированием. В среднем, с момента проявления заболевания (чаще при лабораторно-инструментальной диагностике) до развития фиброза и цирроза печени или необходимости решения вопроса о трансплантации органа проходит не более 4-х лет.
Диагностика ДЛКЛ затруднена ввиду разнообразия клинический проявлений, однако, к счастью, существует простой, точный и БЕСПЛАТНЫЙ тест, позволяющий подтвердить данное заболевание. Для верификации диагноза ДЛКЛ проводится определение активности фермента ЛКЛ в пятнах высушенной крови и при обнаружении снижения активности фермента проводят ДНК-диагностику для поиска мутаций гена LIPA.
Буквально до недавнего времени для терапии ДЛКЛ была доступна лишь симптом - модифицирующая терапия: препараты для снижения холестерина, диета с низким содержанием жира, трансплантация печени, трансплантация гемопоэтических стволовых клеток.
В терапии младенцев с быстропрогрессирующей ранней формой ДЛКЛ (болезнью Вольмана) применялось энтеральное и парентеральное питание с низким содержанием жиров, однако какого-либо влияния на смертность данная терапия не оказывала. Низкожировая диета до настоящего времени применяется как у детей так и взрослых с ДЛКЛ, однако этот подход не продемонстрировал достаточную эффективность.
Результаты терапии пациентов с ДЛКЛ гиполипидемическими препаратами в литературе неоднозначны. До настоящего времени остается неясным, оказывает ли применение статинов позитивное влияние на предотвращение развития атеросклероза, риска сердечно-сосудистых осложнений, прогрессирования поражения печени, ассоциированных с ДЛКЛ.
Сравнительно недавно появился новый подход к лечению ДЛКЛ — заместительная ферментная терапия. В России накоплен достаточный опыт диагностики ДЛКЛ. По данным генетических исследований, в российской популяции предполагается, что частота ДЛКЛ в нашей стране может составлять 1: 67 600. Благодаря знаниям врачей, вниманию к заболеваниям печени как у детей, так и у взрослых, возможностям современных лабораторий в настоящее время диагностированы десятки случаев ДЛКЛ. Осведомленность прежде всего врачей об особенностях клинической картины заболевания важна для его своевременной диагностики, поскольку быстрое прогрессирование ДЛКЛ иногда не дает возможности проводить долгий диагностический поиск.